2-Deoxyglucose (2-DG)
Starving glioblastoma at the molecular level
2-Deoxyglucose is a glucose analog that exploits glioblastoma's metabolic Achilles' heel - its absolute dependence on glycolysis. By mimicking glucose and sabotaging its processing, 2-DG creates an energy crisis selectively lethal to tumor cells.
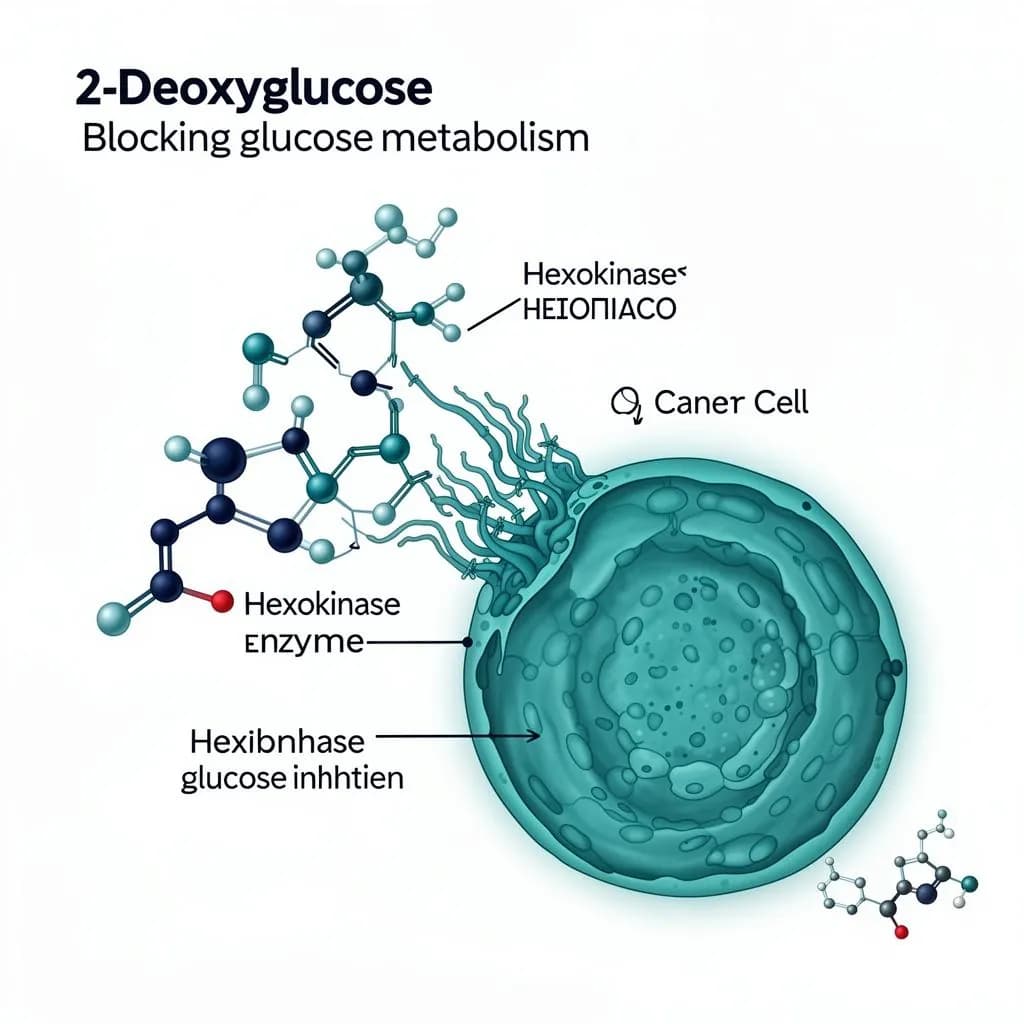
2-DG molecular mechanism
Hexokinase II inhibition at the cellular level
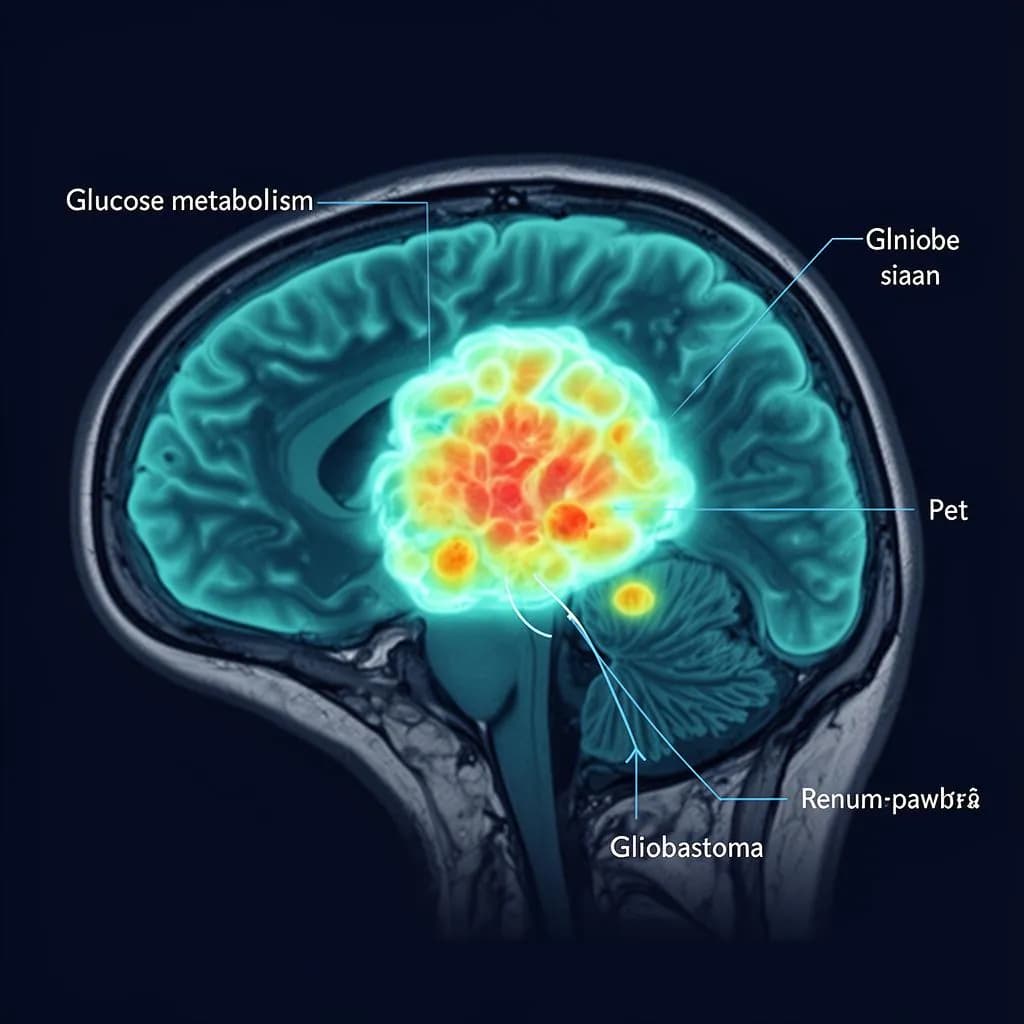
PET scan - glucose uptake
Tumor regions show dramatically elevated glucose metabolism
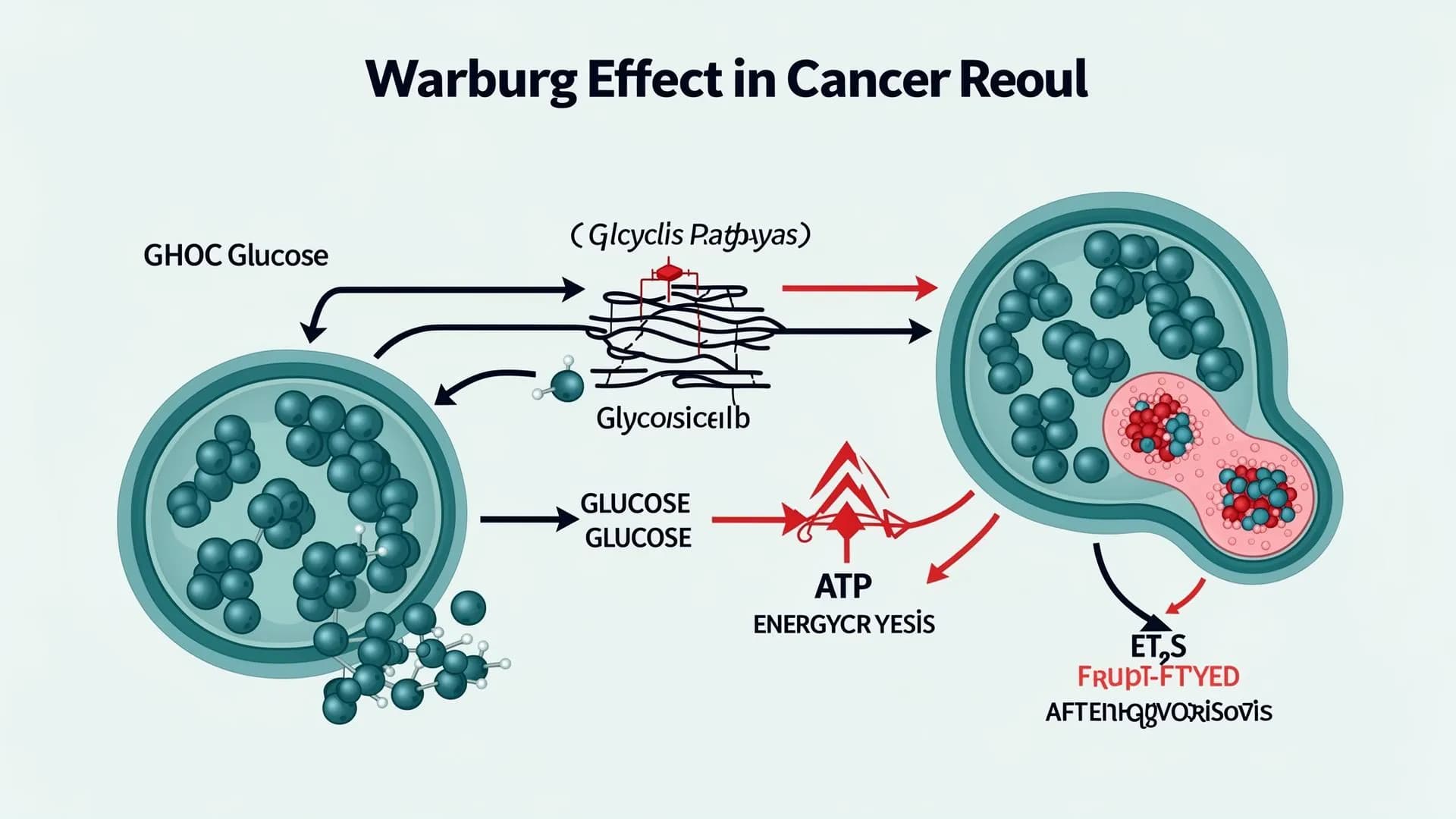
The Warburg effect
How 2-DG exploits cancer's glycolytic dependency
The scientific rationale: why GBM is vulnerable
In 1924, Otto Warburg observed that cancer cells consume glucose at rates 10–100× higher than normal cells, even in the presence of oxygen - a phenomenon now called aerobic glycolysis or the Warburg effect.
200×
Higher glucose uptake in GBM vs. normal brain tissue (PET imaging)
GLUT1/3
Glucose transporters massively overexpressed in GBM - 2-DG exploits this for selective uptake
HK-II
Hexokinase II - the primary target of 2-DG - is 100× overexpressed in GBM cells
GBM has among the highest glycolytic rates of all human cancers. That metabolic addiction fuels rapid growth, and it creates a real vulnerability: block glycolysis, and the tumor starves while normal cells - which use oxidative phosphorylation - remain unaffected.
Molecular mechanisms of action
2-DG attacks GBM through six distinct and synergistic molecular pathways:
Hexokinase II inhibition
2-DG is phosphorylated by hexokinase II to 2-DG-6-phosphate, which cannot be further metabolized. It accumulates intracellularly and competitively inhibits hexokinase II - the first rate-limiting enzyme of glycolysis - effectively blocking glucose utilization at the very first step.
Glycolysis shutdown
By blocking hexokinase II and inhibiting phosphoglucose isomerase, 2-DG halts glycolytic flux. GBM cells - which depend on aerobic glycolysis (the Warburg effect) for >60% of their ATP - experience acute energy crisis, while normal neurons, which rely on oxidative phosphorylation, are largely spared.
ER stress & unfolded protein response
2-DG disrupts N-linked glycosylation in the endoplasmic reticulum by acting as a mannose mimetic. This triggers the unfolded protein response (UPR) via PERK, IRE1α, and ATF6 pathways, leading to CHOP-mediated apoptosis - a mechanism particularly lethal in GBM stem cells.
Autophagy induction
Energy deprivation caused by 2-DG activates AMPK and inhibits mTORC1, triggering autophagy. In GBM, prolonged autophagy under glucose deprivation transitions from protective to cytotoxic - autophagic cell death becomes an additional killing mechanism.
Radiosensitization
2-DG depletes intracellular glutathione and thioredoxin - key antioxidant defenses. This renders GBM cells acutely sensitive to radiation-induced reactive oxygen species (ROS). Phase I/II trials (ACT-GBM-01) demonstrated that 2-DG + radiation improved median survival without increasing toxicity.
Anti-angiogenic & immune effects
2-DG downregulates HIF-1α and VEGF expression under hypoxic conditions, reducing tumor neovascularization. It also modulates the tumor microenvironment by reducing lactate secretion, which restores T-cell cytotoxicity and reverses immunosuppressive polarization of tumor-associated macrophages.
Press-Pulse integration: 2-DG as the metabolic pulse
Within Dr. Thomas Seyfried's Press-Pulse framework, 2-DG is the acute "pulse" - a targeted strike against tumor metabolism during a state of chronic metabolic stress.
Ketogenic diet
Chronic calorie-restricted ketogenic diet lowers blood glucose to 55–65 mg/dL and elevates blood ketones to 3–6 mmol/L, creating sustained metabolic pressure on tumor cells that cannot utilize ketone bodies.
2-DG administration
Acute 2-DG doses (250 mg/kg oral, timed with radiation) block residual glucose utilization, creating a 'metabolic double-hit' - glucose supply is restricted by diet, and glucose processing is blocked by 2-DG.
Combined effect
Together, the Press (diet) + Pulse (2-DG) creates a metabolic environment incompatible with GBM survival while ketone bodies nourish and protect normal brain tissue - a therapeutic window unique to metabolic therapy.
Clinical & preclinical evidence
Peer-reviewed studies supporting the use of 2-DG in glioblastoma:
Dwarakanath et al., 2009
Clinical TrialJournal of Cancer Research & Therapeutics
Phase I/II trial: 2-DG (250 mg/kg) combined with radiation in GBM patients showed improved median survival with manageable side effects. No dose-limiting toxicity was observed.
Raez et al., 2013
Clinical TrialCancer Chemotherapy and Pharmacology
Phase I dose-escalation study demonstrated that oral 2-DG is safe up to 63 mg/kg/day. Serum glucose levels were not significantly altered, confirming selective tumor targeting.
Stein et al., 2010
Imaging StudyNeuro-Oncology
2-DG selectively accumulates in GBM tissue at 3–5× higher concentrations than surrounding brain parenchyma, as confirmed by 18F-FDG PET imaging - exploiting the tumor's elevated glucose transport (GLUT1/GLUT3 overexpression).
Zhang et al., 2014
PreclinicalMolecular Cancer Therapeutics
2-DG synergizes with temozolomide in MGMT-unmethylated GBM cell lines by depleting NAD+ pools through PARP activation, overcoming TMZ resistance - a major clinical challenge.
Maher et al., 2004
PreclinicalMolecular Cancer Therapeutics
In glioma xenograft models, 2-DG (500 mg/kg) reduced tumor volume by 45% as monotherapy and 78% when combined with radiation, with complete responses in 20% of combination-treated animals.
Safety & tolerability profile
Clinical trials have established 2-DG as well-tolerated at therapeutic doses. The key safety findings:
Mild hypoglycemia-like symptoms at high doses (>45 mg/kg)
Transient QTc prolongation at doses >63 mg/kg (reversible)
Fatigue, sweating, and dizziness in ~15% of patients
No hepatotoxicity, nephrotoxicity, or myelosuppression observed
Normal brain cells unaffected due to oxidative metabolism
Key advantage: Unlike conventional chemotherapy, 2-DG does not cause myelosuppression, liver toxicity, or nephrotoxicity. Its selectivity for high-glycolytic cells means normal brain tissue is minimally affected - a critical advantage for a CNS therapeutic.
How we use 2-DG in our protocol
Dosing strategy
Oral 2-DG at 250 mg/kg, administered in a fasted state to maximize tumor uptake. Timed 30–45 minutes before radiation sessions for maximum radiosensitization. Dose is individualized based on patient weight, GKI index, and metabolic response.
Monitoring
Blood glucose and ketone levels tracked in real-time via continuous glucose monitoring. GKI index maintained at 1.0–2.0 during 2-DG administration. ECG monitoring for QTc at higher doses. Serial MRI with metabolic imaging (MR spectroscopy) to assess tumor metabolic response.
Combination partners
Combined with ketogenic diet (press), radiation therapy (radiosensitization), DON (glutamine blockade for dual-pathway metabolic shutdown), and HBOT (hyperbaric oxygen to amplify oxidative stress in glycolysis-impaired tumor cells).
Patient selection
Ideal candidates identified through FDG-PET (confirming high glycolytic activity), molecular profiling (MGMT status, IDH mutation), and drug sensitivity testing on patient-derived tumor cells. Not all GBMs are equally glycolytic - we test, then treat.